Approche scientifique
Origin of Precambrian Banded Iron Formations (BIFs) : Early Oxidant-Producing Photosynthesis Outside A Locally Acidified Sedimentary Environment – A Synthesis
Leo J. Christiaens and Christophe F. Meunier
Abstract
This study of Precambrian BIF origin presents a few postulates, which concern the species of dissolved iron available in the sedimentary environment, and a novel line of thought. Which concerns the 13C depleted carbonate species, with which the sedimentary environment was – locally – supplied. These carbonate species, which were (locally) added to the carbonate species of atmospheric provenance, may have been produced somewhere outside the sedimentary environment. In a stromatolite-housing coastal area, for instance, where carbon dioxide was generated by the anaerobic decay of dead microorganisms. Moreover, carbon monoxide and hydrocarbon, which were also brought forth by the anaerobic decay of dead microorganisms, may have been oxidized – by an oxidant which was most likely not free oxygen – whilst they (and the forementioned CO2) were transported from the said coastal area to the sedimentary environment. The origin of BIF units which include not only iron oxide and chert, but also iron carbonate and iron silicate, probably goes back to sedimentary environments which were supplied with abundant 13C depleted carbonate species.
Purposeful interpretation of available data results in the development of a mineralogically pertinent model of the sedimentary environment. Which was supplied with: (1) (abundant) reactive oxygen species (viz. H2O2), by a dispersed surface flux in a coastal current; (2) (abundant) dissolved (13C depleted) carbonate species, by a dispersed bottom flux in the said coastal current; (3) alkaline marine basin water; (4) Fe(OH)2 and H4SiO4-rich continental solutions; and (5) abundant H2 and CO2, by a «bubble» atmosphere, with the sedimentary environment being part of a subglobal system.
The H2O2 plausibly was a product of prime photosynthesis (12C was preferentially incorporated into organic compounds), which used H2 to reduce CO2.
The synthesis made in this study gives insight into early geotectonics. As a result of which BIFs are poor in aluminum.
Introduction
BIF units, 50 to 600 m thick, occur around the world, in rock records older than 1.8 billion years (Trendall and Blockley, 1970; Beukes, 1973; Goldich, 1973; Gole and Klein, 1981; Klein, 2005). BIFs represent chemical sediments. They typically consist of iron mineral layers which alternate with bands of cryptocrystalline quartz (chert). In the best preserved BIF units, the mineralogy of iron includes various oxides (magnetite, hematite), carbonates (siderite, ankerite) and silicates (greenalite, minnesotaite, riebeckite, stilpnomelane). Iron sulfide (pyrite) is a minor occurrence. It is generally agreed that most of the forementioned minerals do not represent primary products of chemical sedimentation, and that, instead, they are the result of diagenetic and metamorphic overprinting of thermodynamically unstable compounds (Klein and Bricker, 1977). Ill-defined iron oxyhydroxide, for instance, is what the oxidation of dissolved iron produces today, the oxidizing agent being biologically generated free oxygen (O2). There are, however, no data to support that early biologic activity was directly involved in BIF precipitation (Klein, 2005; Posth et al., 2013). Yet, the origin of BIFs goes back to a complex interplay of processes, notably biospheric ones (Bekker et al., 2010).
If a synthesis has to be made to sort this out, how is it to be made ? That is what this paper is all about.
Postulates
Postulate 1: each major group of BIF minerals points to a particular species of dissolved iron in the sedimentary environment. That is to say: the oxides point to Fe(OH)2, the carbonates to FeOH+, and the silicates to Fe2+.
Postulate 2: each species of dissolved iron had a distribution of its own within the sedimentary environment.
Postulate 3: the arrangement of the different distributions was such that local coprecipitation of different minerals/mineral precursors was inevitable.
Postulate 4: the dissolved iron species released by the reduction of oxidized iron compounds, was Fe2+.
Postulate 5: the iron species the sedimentary environment was being supplied with, was Fe(OH)2.. This implies that local reduction of oxidized iron compounds was needed for Fe2+ to appear, and that local acidification of the sedimentary environment was required for FeOH+ perhaps even Fe2+ to be available.
Local acidification of the sedimentary environment
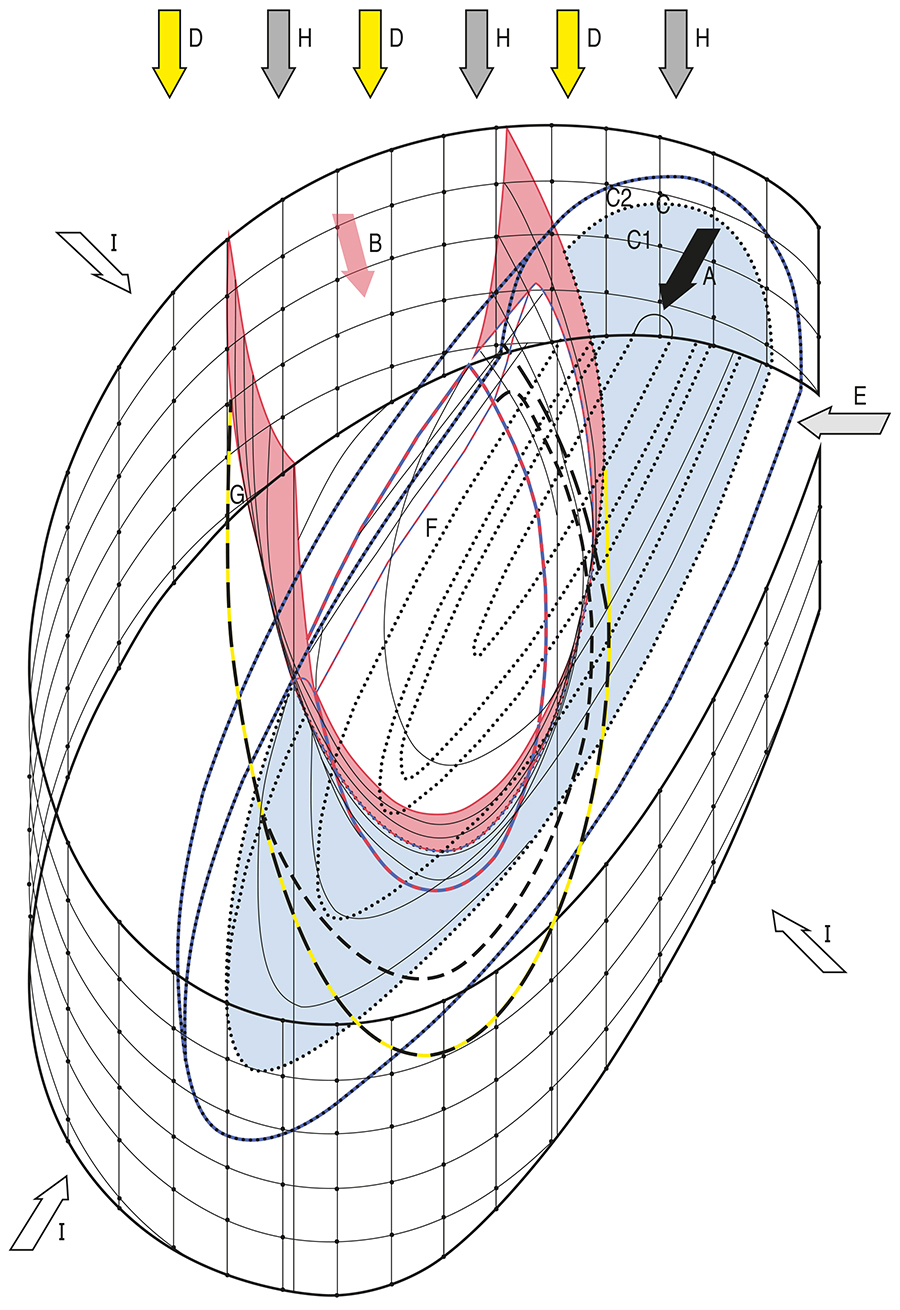
Siderite and ankerite in Early Proterozoic BIFs are depleted in the heavier carbon isotope 13C, relative to dolomites and limestones (Becker and Clayton, 1972; Perry and Tan, 1972; Perry and Ahmad, 1981; Baur et al., 1985; Beukes et al., 1990). This indicates that, locally, the sedimentary environment was provided with carbonate species not only of atmospheric, but also of biospheric provenance (Fig. 1, A). Carbonate of biospheric provenance need not have been produced by the oxidation of organic matter under aerobic conditions. It may, instead, have been generated by the anaerobic decay of organic matter. And, possibly, by the oxidation, somewhere outside the sedimentary environment, of carbon monoxide (CO) and hydrocarbon also released by that decay.
This line of thought implies that somewhere outside the sedimentary environment, there were primitive microorganisms which lived and died. This line of thought also implies that the living ones most likely produced the substance which oxidized the CO, and the hydrocarbon, set free by the anaerobic decay of the dead ones. Anaerobic meaning that the produced oxidant was by far not so widely dispersed as O2 would have been (consequently: is it justified to preclude that hematite and magnetite represent primary products of sedimentation?).
This line of thought further implies that if the oxidizing agent was a noxious substance (H2O2, for instance), the living microorganisms needed the assistance of something which enabled them to immediately get rid of it. A marine (conceivably a coastal) current, for instance. The top of which entrained the oxidizing agent. The bottom of which entrained CO2, CO and hydrocarbon. The possibility of H2O2 being the oxidizing agent is to be further explored, in forthcoming discussions of strong mid-oceanic volcanic activity, and of abundant H2 being available for the reduction of CO2.
To examine the potential of the proposed angle of incidence, a few educated guesses are needed. The first one being that on the way to the sedimentary environment, the oxidant was dispersed, so that the oxidant flux could penetrate the bottom flux. Making the oxidant react with CO and hydrocarbon. The thus produced carbon dioxide (CO2) reacted with water (H2O) to produce carbonic acid (H2CO3). Which was abundant, only if there was widespread interpenetration of the (enlarged) oxidant flux and the bottom flux. That is to say: the sooner (or the more often) the oxidant flux (Fig. 1, B) and the bottom flux (Fig. 1, A) diverged, the less H2CO3 was produced.
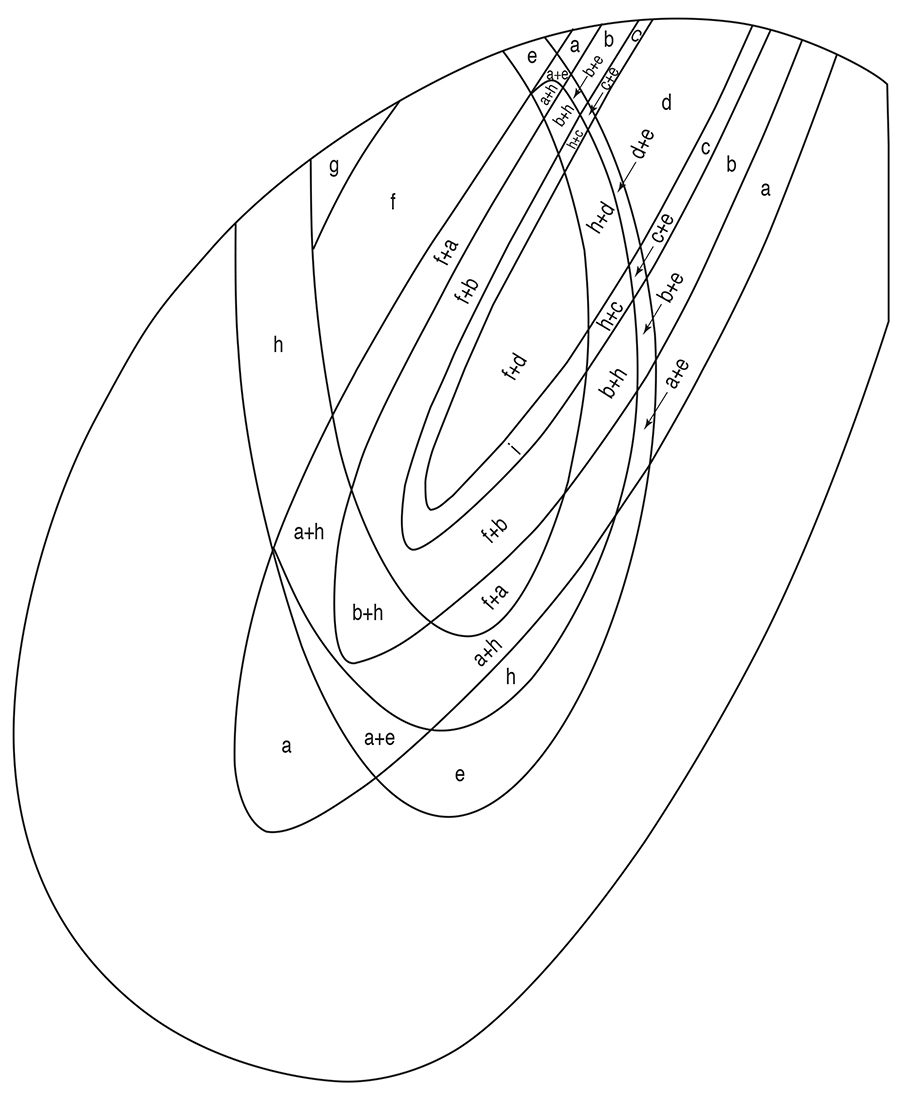
The second educated guess is that the H2CO3-transporting bottom flux and the surrounding marine current waters exchanged H+ and OH- ions and that, therefore, the sedimentary environment was supplied with abundant HCO3- ions. Which reacted with FeOH+ ions. These FeOH+ ions were locally available, as a result of the acidification of the Fe(OH)2-rich solutions provided to the sedimentary environment (Fig. 1, C). The reaction produced FeCO3, siderite (if primary; Fig. 2, a):
FeOH+ + HCO3- => FeCO3 + H2O (1)
The third educated guess is that there where the sedimentary environment was sufficiently rich in H+ (H3O+, actually) ions, FeOH+ ions were partially dissociated. And that they reacted with silicic acid, H4SiO4 (which was also provided to the sedimentary environment), and with OH- ions taking part in the above discussed exchange of H+ and OH- ions. The reaction yielded a thermodynamically unstable iron silicate, Fe3Si4O10(OH)2.10 H2O:
3 Fe2+ + 3 OH- + 4 H4SiO4 + 3 OH- => Fe3Si4O10(OH)2.10 H2O (2)
Which conceivably was the precursor of chert (SiO2; Fig. 2, b). During diagenesis, weakly bound H2O may have been squeezed out, causing the disruption of the structure and the loss of the constituent Fe2+ and OH-:
Fe3Si4O10(OH)2.10 H2O => 8 H2O + 3 Fe2+ + 6 OH- + 4 SiO2 (3)
Elsewhere in the diagenetic (or metamorphic) environment, chert precursor may have been in contact with solutions rich in Fe2+ ions. So rich in Fe2+ ions that constituent Fe2+ and OH- of the chert precursor were effectively prevented from leaving. Dehydration-induced restructuring produced minnesotaite, Fe3Si4O10(OH)2 (Fig. 2, c):
Fe3Si4O10(OH)2.10 H2O => Fe3Si4O10(OH)2 + 10 H2O (4)
Coprecipitated chert precursor and aluminum hydroxide, Al(OH)3, may have reacted with each other, in the diagenetic (or metamorphic) environment, to produce stilpnomelane, (approximately) Fe6Si7AlO20(OH)3 (Fig. 2, d):
2 Fe3Si4O10(OH)2.10 H2O + Al(OH)3 => Fe6Si7AlO20(OH)3 + H4SiO4 + 20 H2O (5)
Aluminum in stilpnomelane indicates that the bottom flux was locally sufficiently acid to transport some aluminum in solution. Aluminum in stilpnomelane also indicates that in the sedimentary environment, the bottom flux was made progressively less acid, so that insoluble Al(OH)3 was locally precipitated.
Early oxidant-producing photosynthesis
The fourth educated guess is that Fe6Si4O10(OH)8, greenalite (if primary; Fig. 2, e) appeared – somewhere in the sedimentary environment – as a result of the reaction of freshly produced chert precursor with dissolved Fe(OH)2:
Fe3Si4O10(OH)2.10 H2O + 3 Fe(OH)2 => Fe6Si4O10(OH)8 + 10 H2O (6)
The reaction which imperatively preceded (6), produced chert precursor. For this reaction to take place, Fe2+ and OH- ions were needed, cf. equation (2). The presence of dissolved Fe(OH)2 implying the shortage of FeOH+ ions, dissociation of FeOH+ could not have been the source of these Fe2+ and OH- ions. It probably was the reduction of oxidized iron compounds which (cf. postulate 4) set free the Fe2+ ions; the OH- ions being provided most likely by waters which, in the sedimentary environment, were situated beyond the reach of strong acidification. The reaction which imperatively preceded (6), can be symbolized by an equation like:
3 Fe2+ + 6 OH- + 4 H4SiO4 => Fe3Si4O10(OH)2.10 H2O (7)
Here, the line of thought being developed in the present study of BIF origin, shows that there was a reducing agent in the sedimentary environment. Which was not organic matter. A reducing agent which (prediction potential is being tested), outside the sedimentary environment, may have been used, by microorganisms, for the reduction of CO2.
Volcanic and hydrothermal activity were of prime importance to the origin of BIFs (Jacobsen and Pimentel-Klose, 1988; Bau and Möller, 1993; Isley, 1995; Isley and Abbot, 1999; Hamade et al., 2003). Therefore, the reductant – hydrogen gas (H2), most likely – may have been available, on a subglobal scale, in a bubble of hot (lower) atmosphere. A bubble which was enclosed within the global atmosphere, which was cold, due to low solar luminosity in Precambrian days (Sagan and Mullen, 1972; Kasting and Ackerman, 1986). A bubble which was separated from the global atmosphere by a sharp drop in temperature (which prevented mixing from readily taking place). A bubble which, possibly with the help of coeval bubbles present elsewhere, prevented the Earth from being covered by ice from pole to pole (Sellers, 1969, 1973; Kirshvink, 1992; Kopp et al., 2005).
The fifth educated guess is that primitive microorganisms had discovered how, on Earth, it was possible to make happen reactions which produce organic carbon, (approximately) C2nH4nO2n, and which are symbolized by the equations (8) and (9):
n CO2 + 2n H2 + 2n H2O => n CH4 + 2n H2O2 (8)
n CH4 + n CO2 => C2nH4nO2n (9)
The total reaction for early oxidant-producing photosynthesis would have been represented by the equation:
2n CO2 + 2n H2 + 2n H2O => C2nH4nO2n + 2n H2O2 (10)
Meaning that the reducing agent was H2, and that the primitive microorganisms had to immediately get rid of H2O2.
The H2O2 which was not consumed by the oxidation of CO and hydrocarbon, was brought to the sedimentary environment. By a marine current. Conceivably, a coastal current. Therefore, the sedimentary environment may have been a shallow-water environment. Yet, the lack of current and wave-generated structures in BIFs (Morris and Horwitz, 1983; Klein and Beukes, 1992) is believed to indicate that BIFs were precipitated at water depths of more than 200 m. The present study shows, however, that for a BIF unit to be deposited, a coastal current was needed. Therefore, the lack of current-generated structures in BIFs probably points to a coastal current which was decelerated, as a result of its being mixed – in the sedimentary environment – with, most likely, continental outflow. Consequently, continental solutions may have been the ones which supplied the sedimentary environment with abundant dissolved Fe(OH)2 (Fig. 1, E).
Rare earth element (REE) signatures in BIFs (Dymek and Klein, 1988; Klein and Beukes, 1989) are believed to indicate that the marine basin waters with which the sedimentary environment was provided, transported REEs which were delivered by a deep-marine hydrothermal source. It should be pointed out, however, that these marine basin waters did not necessarily also bring abundant dissolved iron to the sedimentary environment. Plausibly, REE signatures in BIFs show that, locally, the Precambrian sea floor stood higher than it did elsewhere (and much higher, presumably, than the sea floor stands, on average, at present). Locally, that is to say: there where the heat the inner Earth produced, was very abundant. And where, in the heart of a mid-oceanic ridge, oceanic crust was generated (much) more rapidly than it is today (Allègre, 1982). And where, therefore, the mid-oceanic ridge emerged (or nearly so) from the ocean. So that the («bubble») atmosphere received abundant heat and gases, and the upper ocean, abundant hydrothermal solutions. Therefore, REE signatures in BIFs do not necessarily point to a relatively deep-marine sedimentary environment for BIF origin. And winds in a «bubble atmosphere» may not have been sufficiently strong, for waves to be generated which, in a shallow ocean, would have been capable of leaving their «footprints» in the sediments of the most shallow of sea waters.
In the sedimentary environment
Locally in the sedimentary environment, viz. there where H2O2 was available (Fig. 1, F), H2O2 reacted with dissolved Fe(OH)2 to produce Fe3O4, magnetite (Fig. 2, f), and water:
3 Fe(OH)2 + H2O2 => Fe3O4 + 4 H2O (11)
Or to produce Fe2O3, hematite (Fig. 2, g) and water:
2 Fe(OH)2 + H2O2 => Fe2O3 + 3 H2O (12)
The produced iron oxide settled down. The iron oxide which, whilst settling down, left the portion of the sedimentary environment where H2O2 was available, was reduced by the H2 which was supplied by the «bubble atmosphere» to subaerial waters (Fig. 1, G):
Fe3O4 + H2 + 6 H+ => 3 Fe2+ + 4 H2O (13)
Or:
Fe2O3 + H2 + 4 H+ => 2 Fe2+ + 3 H2O (14)
That is to say: only there where H+ ions were in sufficient supply. According to what was hitherto taken into consideration, this was the case only in the acidified portion of the sedimentary environment. Local interpenetration of the acidified portion and the H2O2-receiving portion, is evident from the observed coexistences of magnetite and siderite, for instance.
The released Fe2+ ions reacted with dissolved H4SiO4 to produce chert precursor (Fig. 2, h), cf. the above discussion on that subject, equation (7) indicating that the reaction could take place, only in waters which were situated beyond the reach of strong acidification. Greenalite appeared only there where Fe(OH)2 was available, cf. equation (6).
Let it be reminded, however, that – to distinguish between local and overall acidification of the sedimentary environment – input of carbonate species of atmospheric
provenance has not yet been appropriately accounted for.
In the diagenetic (or metamorphic) environment
Oxidized iron is an essential constituent not only of hematite and magnetite, but also of riebeckite, an iron silicate. The composition of which, Na2Fe(II)3Fe(III)2Si8O22(OH)2, indicates that the precursor substances were (locally coexistent) chert precursor and magnetite (or hematite). Given that local interpenetration of the acidified portion and the H2O2-receiving portion of the sedimentary environment was possible, the chert precursor may have been chert precursor which had been precipitated through the strongly acidified portion of the sedimentary environment. The solutions the coexistent chert precursor and magnetite were in contact with – in the diagenetic (or metamorphic) environment, of course – may therefore have been rich in Fe2+ ions. So rich in Fe2+ ions that constituent Fe2+ would have been effectively prevented from leaving the chert precursor (cf. the above discussion on that subject). So rich that dehydration-induced restructuring would have produced minnesotaite, cf. equation (4). If magnetite had not been there. But, magnetite was there:
2 Fe3Si4O10(OH)2.10 H2O + Fe3O4 + 2 Na+ + 6 H+ => Na2Fe(II)3Fe(III)2Si8O22(OH)2 + 4 Fe2+ + 24 H2O (15)
And riebeckite arose (Fig. 2, i).
In an ongoing synthesis: the sixth educated guess
In the Precambrian, a particularly productive, highstanding mid-oceanic ridge system was at work, an unknown distance away from the area where BIFs were being deposited.
As a result, a small, thin continent had, some unknown time earlier, been pushed back and had collided with a big, thick continent. The sea which at the outset separated the small continent from the big one, got enclosed and became an inland basin. Which collected the solutes released by the rocks which showed on the surface of the vast continental hinterland.
Yet, the thin and the thick continent did not get firmly welded. Had it been possible to measure the effective elastic strength of the «craton», low values would have been obtained on the side of the small continent, and large values on the side of the big continent. Interestingly: the elastic thickness of the continental lithosphere is still strongly bi-modal (Grotzinger and Royden, 1990; Watts, 1992).
Again and again, the weld gave in. Why? Was the oceanic plate moving? Did it therefore frequently transmit an impulse to the small continent? Put the case that it did, was it responsible for BIF strata being broken, indeed for fragments being rounded? That is to say: was the oceanic plate also responsible for the origin of granular iron formation?
On the «craton» side of the small continent, the weld gave in. Again and again, earthquakes broke the surface rocks up. Drainage therefore was effective: the removal of solutes set free by continental weathering, was not hindered by soil formation.
The «bubble atmosphere» was most likely rich not only in H2, but also in CO2 (Fig. 1, H); cf. the discussion on that subject in the conclusion. Subaerial weathering therefore resulted in the complete dissociation of surface rocks into dissolved substances, which were transported to the inland basin. Which, given the poor welding, conceivably would have been emptied, if the hole in its bucket had not been stopped. Stopped with insoluble aluminum hydroxide, Al(OH)3: BIFs are poor in aluminum (Klein, 1978). Suggesting that the inland basin waters were less acid than the inflowing solutions.
Overflow of the inland basin supplied the sedimentary environment with Fe(OH)2 and H4SiO4-rich solutions.
Conclusion
The Precambrian BIF record does not speak for itself, and the wealth of processed data has not yet made current theory of BIF origin (Cairns-Smith, 1978; Braterman et al., 1983; Klein, 2005; Holland, 2006; Bekker et al., 2010; Rasmussen et al., 2015) rise above controversy.
Therefore, and to summarize the above discussions: the global atmosphere did not play a key role in BIF origin. Subglobal «hot bubble» atmospheres did. As a result of the very productive volcanic-exhalative activity of highstanding mid-oceanic ridge systems.
Free oxygen did not play a key role in BIF origin. Hydrogen peroxide did. It may have been produced by early oxidant-producing photosynthesis
Upwelling currents from the deep ocean did not play a key role in BIF origin. Coastal currents did. They removed H2O2 from oxidant-producing primitive microorganisms. H2O2 which they brought to the sedimentary environment that, therefore, was locally oxidizing. Coastal currents also brought acid carbonate species to the sedimentary environment which, as a consequence, was locally acidified.
Marine basin water (Fig. 1, I) did not supply the sedimentary environment with abundant dissolved iron. Continental solutions did. Because the «bubble atmospheres» were rich in CO2 and H2. So that continental weathering was most effective. Aided as it was by the then, on a subglobal scale ruling geotectonics.
«Bubble atmospheres» provided BIF sedimentary environments with abundant H2. So that freshly produced iron oxide was locally reduced. That is to say, in view of the proved relevancy of the postulates at the beginning of this study: in BIF sedimentary environments, reduction of iron oxide preceded the appearance of (part of the released) chert precursor, and of greenalite.
Interestingly, if oxidized iron in BIFs and associated iron ores was right from the beginning crystalline-textured, primary magnetite or hematite, then it was right from the beginning saturated with O. Consequently, post-depositional oxidation did not take place and, therefore, was unable to delay the Great Oxygenation Event. The GOE benefited most likely from «bubble atmospheres» becoming rarer, less voluminous and poorer in H2 and CO2.
Stilpnomelane, minnesotaite, riebeckite and siderite are conspicuously lacking in BIF units which consist exclusively of iron oxide and chert. This indicates that local acidification of the sedimentary environment did not always apply. And that the background, overall acidification of the said environment, by carbonate species of atmospheric provenance, supplied sufficient H+ ions for magnetite and hematite to be locally reduced, by H2 of atmospheric provenance. Meaning that the «bubble atmosphere» was rich in H2 and in CO2, and that the «bubble ocean» may therefore have been less alkaline that «the» ocean. And that there where future orebodies (made exclusively of iron oxide) were in process of formation, the atmospheric CO2 partial pressure and/or the atmospheric H2 partial pressure had dropped below a critical value.
BIFs are a massive yet very subtle code of the potential which Precambrian environments and dynamics had, for economically important mineral deposits to be brought forth. How does ankerite, how do manganese-rich deposits fit in with the picture? What do they add to it?
What is to be discovered on the hidden side of H2O2-producing photosynthesis?
Hopefully, future research will find out.


References
Allègre, C. J. (1982) Chemical geodynamics. Tectonophys. 81 : 109-132.
Bau, M. and Möller, P. (1993) Rare earth element systematics of the chemically precipitated component in Early Precambrian iron-formations and the evolution of the terrestrial atmosphere-hydrosphere-lithosphere system. Geochim. Cosmochim. Acta 57 : 2239-2249.
Baur, M. E., Hayes, J. M., Studley, S. A. and Walter, M. R. (1985) Millimeter-scale variations of stable isotope abundances in carbonates from banded iron-formations in the Hamersley Group of Western Australia. Econ. Geol. 80 : 270-282.
Becker, R. H. and Clayton, R. N. (1972) Carbon isotopic evidence for the origin of a banded iron-formation in Western Australia. Geochim. Cosmochim. Acta 36 : 577-595.
Bekker, A., Slack, J. F., Planavsky, N., Krapež, B., Hofmann, A., Konhauser, K. O. and Rouxel, O. J. (2010) Iron Formation : the sedimentary product of a complex interplay among mantle, tectonic, oceanic and biospheric processes. Econ. Geol. 105 : 467-508.
Beukes, N. J. (1973) Precambrian iron-formations of Southern Africa. Econ. Geol. 68 : 960-1004.
Beukes, N. J., Klein, C., Kaufman, A. J. and Hayes, J. M., (1990) Carbonate petrography, kerogen distribution, and carbon and oxygen isotope variations in an Early Proterozoic transition from limestone to iron-formation deposition, Transvaal Supergroup, South Africa. Econ. Geol. 85 : 663-690.
Braterman, P. S., Cairns-Smith, A. G. and Sloper, R. W. (1983) Photo-oxidation of hydrated Fe2+ – significance for banded iron formations. Nature 303 : 163-164.
Cairns-Smith, A. G. (1978) Precambrian solution photochemistry, inverse segregation, and banded iron formations. Nature 276 : 807-808.
Dymek, R. F. and Klein, C. (1988) Chemistry, petrology, and origin of banded iron-formation lithologies from the 3800 Ma Isua supracrustal belt, west Greenland. Precambrian Res. 39 : 247-302.
Goldich, S. S. (1973) Ages of Precambrian iron-formations. Econ. Geol. 68 : 1126-1135.
Gole, M. J. and Klein, C. (1981) Banded iron-formations through much of Precambrian time. J. Geol. 89 : 169-183.
Grotzinger, J. and Royden, L. (1990) Elastic strength of the Slave craton at 1.9 Gyr and implications for the thermal evolution of the continents. Nature 347 : 64-66.
Hamade, T., Konhauser, K. O., Raiswell, R., Goldsmith, S. and Morris, R. C. (2003) Using Ge/Si ratios to decouple iron and silica fluxes in Precambrian banded iron formations. Geology 31 : 35-38.
Holland, H. D. (2006) The oxygenation of the atmosphere and oceans. Phil. Trans. R. Soc. B 361 : 903-915.
Isley, A. E. (1995) Hydrothermal plumes and the delivery of iron to banded iron formation. J. Geol. 103 :169-185.
Isley, A. E. and Abbot, D. H. (1999) Plume-related mafic volcanism and the deposition of banded iron formation. J. Geophys. Res. 104 : 15461-15477.
Jacobsen, S. B. and Pimentel-Klose, M. R. (1988) A Nd isotopic study of the Hamersley and Michipicoten banded iron formations : the source of REE and Fe in Archean oceans. Earth Planet. Sci. Lett. 87 : 29-44.
Kasting, J. F. and Ackermann, T. P. (1986) Climatic consequences of very high carbon dioxide levels in the Earth’s early atmosphere. Science 234 : 1383-1385.
Kirshvink, J. (1992) Late Proterozoic low-latitude global glaciation. In : The Proterozoic Biosphere : A Multidisciplinary Study (Eds J. W. Schopf and C. Klein). Cambridge University Press, Cambridge.
Klein, C. (1978) Some aspects of the sedimentary and diagenetic environment of Proterozoic banded iron-formations – a reply. Econ. Geol. 73 : 1371-1373.
Klein, C. (2005) Some Precambrian banded iron formations (BIFs) from around the world : their age, geologic setting, mineralogy, metamorphism, geochemistry, and origin. Am. Mineral. 90 : 1473-1499.
Klein, C. and Beukes, N. J. (1989) Geochemistry and sedimentology of a facies transition from limestone to iron-formation desposition in the early Proterozoic Transvaal Supergroup, South Africa. Econ. Geol. 84 : 1733-1774.
Klein, C. and Beukes, N. J. (1992) Time distribution, stratigraphy, and sedimentologic setting, and geochemistry of Precambrian Iron-Formations. In : The Proterozoic Biosphere : A Multidisciplinary Study (Eds J. W. Schopf and C. Klein). Cambridge University Press, Cambridge.
Klein, C. and Bricker, O. P. (1977) Some aspects of the sedimentary and diagenetic environment of Proterozoic banded iron-formation. Econ. Geol. 72 : 1457-1470.
Kopp, R. E., Kirshvink, J. L., Hilburn, I. A. and Nash, C. Z. (2005) The Paleoproterozoic snowball Earth : A climate disaster triggered by the evolution of oxygenic photosynthesis. Proc. Natl. Acad. Sci. USA. 102 : 11131-11136.
Morris, R. C. and Horwitz, R. C. (1983) The origin of the iron-formation-rich Hamersley Group of Western Australia – Deposition on a platform. Precambrian Res. 21 : 273-297.
Perry, E. C., Jr, and Ahmad, S. N. (1981) Oxygen and carbon isotope geochemistry of the Krivoy Rog iron formation, Ukranian SSR. Lithos 14 : 83-92.
Perry, E. C., Jr, and Tan, F. C. (1972) Significance of oxygen and carbon isotope variations in early Precambrian cherts and carbonate rocks of southern Africa. Geol. Soc. America Bull. 83 : 647-664.
Posth, N. R., Konhauser, K. O. and Kappler, A. (2013) Microbiological processes in banded iron formation deposition. Sedimentology 60 : 1733-1754.
Rasmussen, B., Krapež, B., Muhling, J. R. and Suvorova, A. (2015) Precipitation of iron silicate nanoparticles in early Precambrian oceans marks Earth’s first iron age. Geology 43 : 303-306.
Sagan, C. and Mullen, G. (1972) Earth and Mars : Evolution of atmospheres and surface temperatures. Science 177 : 52-56.
Trendall, A. F. and Blockley, J. F. (1970) The iron formations of the Precambrian Hamersley Group, Western Australia ; with special reference to the associated crocidolite. W. Austr. Geol. Surv. Bull. 119 : 336 pp.
Watts, A. B. (1992) The effective elastic thickness of the lithosphere and the evolution of foreland basins. Basin Research 4 : 169-178.